Preparing Data
Download the following simulated data files from http://gsaa.unc.edu
1, Gene expression dataset : 1.gct
/gene_expression/400/10.3/1.gct
2, SNP dataset : 1.snp
/snp/400/0.7/1.snp
3, Phenotype labels file for gene expression dataset : pheno_400_exp.cls
/phenotype_label/400/pheno_400_exp.cls
4, Phenotype label file for SNP dataset : pheno_400_snp.cls
/phenotype_label/400/pheno_400_snp.cls
5, Gene set database: gt_20_20.gmt
/gene_set/gt_20_20.gmt
Preparing Program
Download GSAA at http://gsaa.unc.edu. In order to use the simulation function in GSAA, you must choose Simulation as the value for the Species parameter.
Getting Started
Unzip or untar the downloaded program file into a directory. Remember, lib and GSAA.jar must be in the same directory.
Windows user:
To launch GSAA, double click the icon of GSAA.jar file or use command
Java –Xmx1000m –jar full-path/GSAA.jar
Linux and Mac user:
Java –Xmx1000m –jar full-path/GSAA.jar
Parameter –Xmx specifies the amount of memory available to Java. 1000m should be enough for simulated datasets. If you get error message “out of memory”, try to increase 1000m to 2000m or more. GSAA has been successfully used with 20000m on a Linux server for a large GWA dataset and 10000 permutations of phenotype labels.
full_path is the complete path of the GSAA.jar file
Example: Java –Xmx1000m –jar C:/programs/gsaa/GSAA.jar
Loading Data

The icons on the left provide quick access to the most common actions. Typically, each action you select opens a new page in the GSAA window. For example, selecting the Load data icon opens the Load data page.
For real dataset, you can browse and select the gene set database from the Broad website. For simulated dataset, you need to load one of the gene set database files, say gt_20_20.gmt, into the application by Load data page.
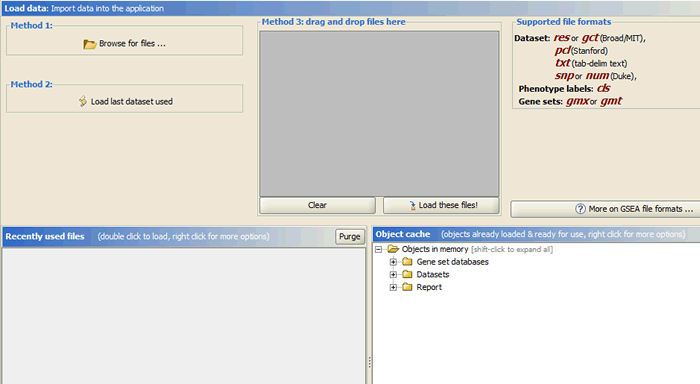
There are several ways to load data:
1. Clicking the Browse for files button will allow you to select files from your file system and load it into GSAA. To select multiple files, use SHIFT-click or CTRL-click.
2. Clicking the Load last dataset used button will load the data used in the most recent analysis.
3. Drag-and-drop the files from a file browser window into the drag-and-drop pane. When the files that you want to load are listed in that pane, click the Load these files button. To remove files from the drag-and-drop pane, click the Clear button.
4. The Recently Used Files pane contains files that you have used previously. Double-click a file to load it.
Specifying Parameters
Click the icon “Run GSAA” to open the GSAA page. Specify parameters before proceeding to run the analysis. There are three categories of parameters in GSAA:
1. Required: Essential parameters which you must specify before the analysis can be run.
2. Basic: Additional parameters with standard defaults. Typically, accepting the defaults is ok. Click Show to see these parameters.
3. Advanced: Parameters that allow control of several more details of the GSAA algorithm and the java implementation. Typically, these do not need to be changed by most users. Click Show to see these parameters.
In this section, parameters are labeled as numbers in Figures
1) Required parameters
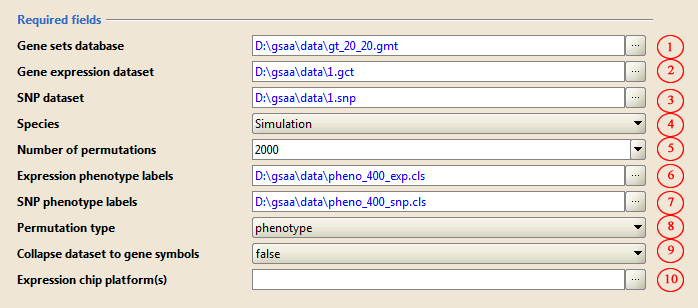
1. Use the ... button open Select one or more gene sets(s) window that lists gene sets in a number of different tabs. For this example, on the GeneMatrix (local gmx/gmt) tab select the gt_20_20.gmt.
2. Use the … button to select gene expression dataset 1.gct from a file browser window.
3. Use the … button to select SNP dataset 1.snp from a file browser window.
4. Use the drop-down list to choose species. For this example, select Simulation from the bottom of the list.
5. Type in or choose the number of permutations to perform. For this example, use 2000.
6. Use the … button to select phenotype label file for gene expression dataset pheno_400_exp.cls from a file browser window.
7. Use the … button to select phenotype label file for SNP dataset pheno_400_snp.cls from a file browser window.
8. Leave the Permutation type parameter set to phenotype.
9. Leave the Collapse dataset to gene symbols parameter set to false.
10. Use the ... to select the chip annotation file that matches the probe identifiers in your expression dataset. For this example, just leave it blank.
2) Basic parameters
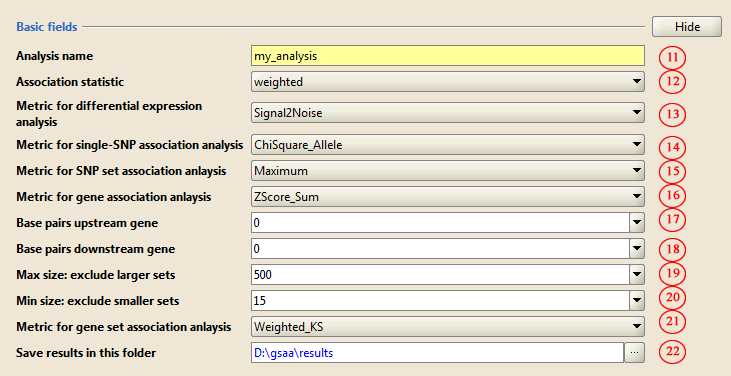
11. Type in a name for the analysis
12-16. Accept the defaults
17. Leave the Base pairs upstream gene parameter set to 0.
18. Leave the Base pairs downstream gene parameter set to 0.
19-21. Accept the defaults.
22. Use the … button to select the directory in which to place output from the analysis
3) Advanced parameters
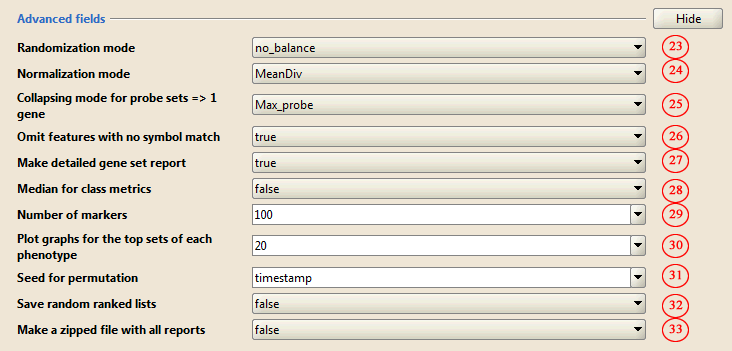
23-33. Accept the defaults
Running GSAA
![]()
Click Run to start the analysis
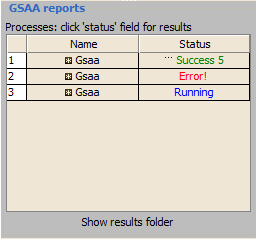
Use the Processes panel at the lower left corner to view the status of analyses run in this session, including the currently running analysis:
1. The blue Running label indicates the currently running analysis. You can click on this label to pause or resume an analysis.
2. If a red Error appears, click on it for a description of the error.
3. When the analysis completes, click the green Success label to display the results in a web browser.