Introduction
Gene Set Association Analysis (GSAA) is a Bioinformatics platform for integrative gene set association analysis of SNP data and microarray gene expression data. GSAA identifies pathways/gene sets significantly associated with a disease or a phenotype through integrating evidence from genome-wide patterns of genetic variation and gene expression variation of two phenotypes.
The software GSAA is a Java based desktop application which implements methods described in
Qing Xiong, Nicola Ancona, Elizabeth R. Hauser, Sayan Mukherjee, Terrence S. Furey. Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome Research. 2012 Feb;22(2):386-97.
Downloading Software
Software GSAA is available for free download at http://gsaa.unc.edu
This download also contains three other software: GSAA-SNP, GSAASeqSP, and GSAASeqGP for gene set association analysis of SNP genotype data and RNA-Seq data.
GSAA can run on any desktop computer (Windows, Mac OS X, Linux etc.) that supports Java7+. Java7+ is available at http://java.sun.com/javase/downloads/index.jsp
Getting Started
Starting GSAA Desktop Application
Unzip or untar the downloaded program file into a directory. Remember, lib and GSAA.jar must be in the same directory.
Windows user:
To launch GSAA, double click the icon of GSAA.jar file or use command
Java –Xmx1000m –jar full-path/GSAA.jar
Linux and Mac user:
Java –Xmx1000m –jar full-path/GSAA.jar
Parameter –Xmx specifies the amount of memory available to Java. If you get error message “out of memory”, try to increase 1000m to 2000m or more. GSAA has been successfully used with 20000m on a Linux server for a large GWA dataset and 10000 permutations of phenotype labels.
full_path is the complete path of the GSAA.jar file
Example: Java –Xmx1000m –jar C:/programs/gsaa/GSAA.jar
When GSAA starts, the main window appears. The main components of the user interface are as follows:
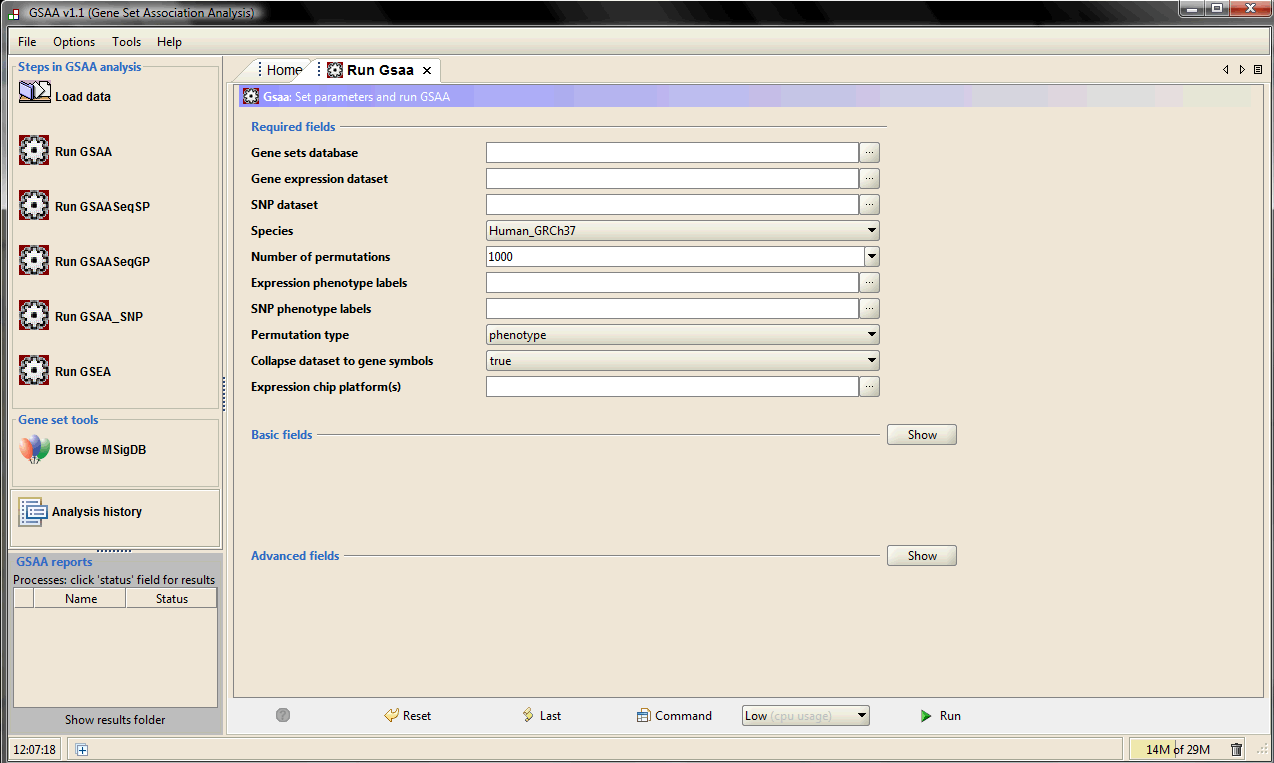
1. The navigation bar on the left, which provides quick access to common GSAA operations.
2. The Processes panel in the bottom left corner, which provides information about the status of your analyses.
3. The main panel on the right, which is used to display dialogs and results. When you start GSAA, the main panel displays the Home page. To open GSAA page, click the icon "Run GSAA", Gsaa tab will appear next to the Home tab. To close the page, click the close (X) icon on the tab.
Exiting GSAA
To exit from GSAA:
1. Click the close (x) button on the top-right corner of the GSAA window.
2. Select File>Exit.
Getting Help
The GSAA web site is your primary source of help for GSAA. It includes the following resources:
1. Documentation. The GSAA documentation includes this User Guide and a Tutorial that walks you through an example analysis based on simulated datasets.
2. Publications. The web site provides a link to the paper describing the algorithms.
If you cannot find the answers to your questions on our web site, contact us at qing.xiong@duke.edu.
Preparing Data Files for GSAA
When you use GSAA, you supply six data files: an expression dataset file, a SNP dataset file, a phenotype labels file for expression dataset, a phenotype labels file for SNP dataset, a gene sets file, and a chip annotations file. The following table lists each type of data file and its valid file formats. All files are tab-delimited ASCII text files; they can be created and edited using any text editor.
| Data File | Content | Format | Source |
|---|---|---|---|
| Expression dataset | Contains features (genes or probes), samples, and an expression value for each feature in each sample. Expression data can come from any source. | res, gct, pcl, or txt | You create the file. |
| SNP dataset | Contains features (SNPs), genomic locations, samples, and a genotype for each feature in each sample. SNP data can come from any source. | snp | You create the file. |
| Phenotype labels | Contains phenotype labels and associates each sample with a phenotype. Only categorical labels are allowed in GSAA. | cls | You create the file. |
| Gene sets | Contains one or more gene sets. For each gene set, gives the gene set name and list of features (genes or probes) in that gene set. | gmx or gmt | You use the files on the Broad ftp site, export gene sets from the Molecular Signature Database (MSigDb) or create your own gene sets file. |
| Chip annotations | Lists each probe on a DNA chip and its matching HUGO gene symbol. Optional for the gene set association analysis. | Chip | You use the files on the Broad ftp site, download the files from the GSEA web site, or create your own chip file. |
You can create and edit GSAA files using Excel or any text editor. If you use Excel to create a tab-delimited text file: select File>Save As, enter the file name in quotes to preserve the the file extension (for example, "lung.snp"), and select "Text(Tab delimited)(*.txt)" as the file type. Excel displays a message warning you that your file may contain features that are not compatible with this format and asks if you want to keep the workbook in this format. Click Yes to keep this format. Your file has now been saved. Exit from Excel. When Excel asks if you want to save your changes to this file, select No (you have already saved the file).
In addition, do not use hypens (-) in the file names.
For descriptions and examples of GSEA-related file formats res, gct, pcl, txt, gmx, gmt, and Chip, see GSEA User Guide and GSEA file formats. For GSAA file formats, see below
SNP Data Format (*.snp)
Note: Genotypes must be coded as 0, 1, and 2 representing AA, AB, and BB, respectively. In SNP dataset, you must sort SNPs first by chromosome number from smallest to largest and then by genomic location from smallest to largest for each chromosome. GSAA currently can analyze data for human, mouse and other 23 species. For human, the chromosome X, Y and MT (mitochondrial chromosomes) must be coded as 23, 24, and 25 respectively. For mouse, the chromosome X, Y and MT must be coded as 20, 21, and 22 respectively. For other species, click HERE to see how to code their chromosomes.
The SNP format is a tab delimited file format that describes a SNP dataset. It is organized as follows:
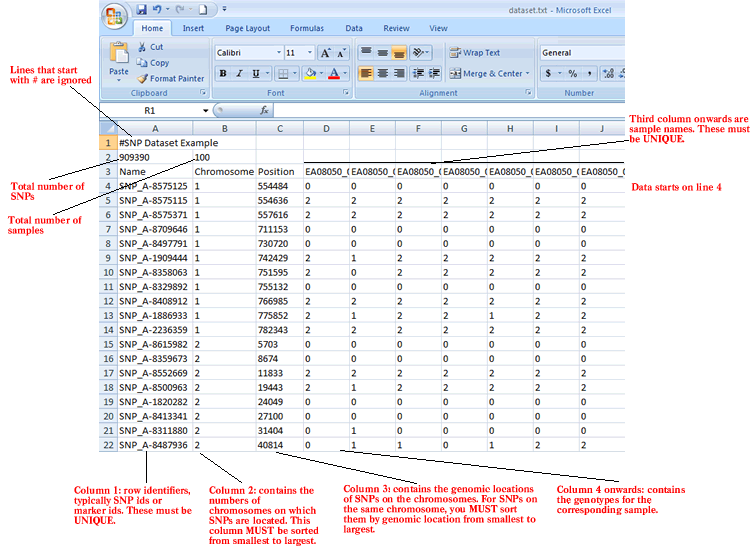
The first line contains comments describing the dataset. The first line must start with #.
Line format: # anything
Example: # lung cancer dataset
The second line contains the number of SNPs and the number of samples.
Line format: (number of SNPs) (tab) (number of samples)
Example: 909390 100
The third line contains a list of identifiers for the samples associated with each of the columns in the remainder of the file.
Line format: Name (tab) Chromosome (tab) Position (tab) (sample 1 name) (tab) (sample 2 name) (tab) ... (sample N name)
Example: Name Chromosome Position EA08050_1 EA08050_2 EA08050_3 EA08050_4 EA08050_5
The remainder of the data file contains data for each of the SNPs. There is one row for each SNP and one column for each of the samples. The number of rows and columns should agree with the number of rows and columns specified on line 2. Each row contains a name, a chromosome number, a genomic location and a genotype for each sample.
Line format: (SNP name) (tab) (chromosome number) (tab) (position) (tab) (col 1 data) (tab) (col 2 data) (tab) ... (col N data)
Example: SNP_A-1909444 1 742429 2 1 2 2 2 2 2 2 1 2 2 1 2 0
Phenotype Data Format (*.cls)
The CLS file format defines categorical phenotype (class or template) labels and associates each sample in the expression or SNP data with a label. Only two phenotypic classes, for example, tumor vs normal, are allowed. The name and label for each class in the expression phenotype labels file must be same as those in the SNP phenotype labels file. However, the number of samples in each class can be different between these two phenotype labels files. We recommend that you use the class of interest, for example tumor, as the first class in the CLS file.
The CLS file format uses spaces or tabs to separate the fields. It is organized as follows:
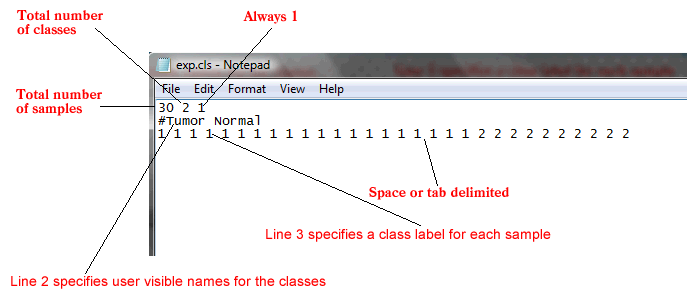
The first line of a CLS file contains numbers indicating the number of samples and number of classes (2). The number of samples should correspond to the number of samples in the associated GCT, RES, or SNP data file.
Line format: (number of samples) (space) 2 (space) 1
Example: 30 2 1
The second line in a CLS file contains a user-visible name for each class. These are the class names that appear in analysis reports. The line should begin with a pound sign (#).
Line format: # (class 1 name) (space) (class 2 name)
Example: #Tumor Normal
The third line contains a class label for each sample. The class label can be the class name, a number, or a text string. The first label used is assigned to the first class named on the second line; the second unique label is assigned to the second class named. (Note: The order of the labels determines the association of class names and class labels, even if the class labels are the same as the class names.) The number of class labels specified on this line should be the same as the number of samples specified in the first line. The number of unique class labels specified on this line should be the same as the number of classes specified in the first line.
Line format: (sample 1 class) (space) (sample 2 class) (space) ... (sample N class)
Example: 1 1 1 ... 2 2
Loading Data
Click the icon “Load data” to open the Load data page.
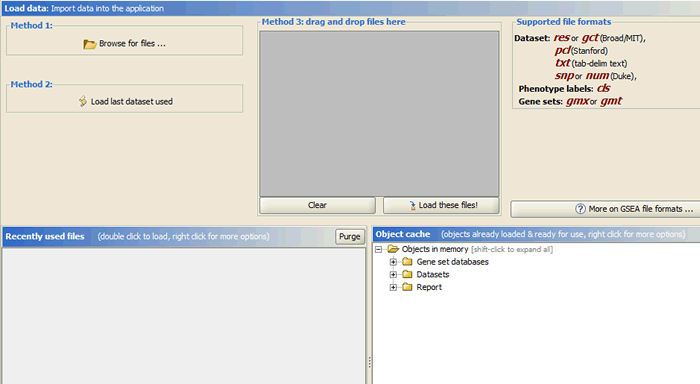
There are several ways to load data:
- Clicking the Browse for files button will allow you to select files from your file system and load it into GSAA. To select multiple files, use SHIFT-click or CTRL-click.
- Clicking the Load last dataset used button will load the data used in the most recent analysis.
- Drag-and-drop the files from a file browser window into the drag-and-drop pane. When the files that you want to load are listed in that pane, click the Load these files button. To remove files from the drag-and-drop pane, click the Clear button.
- The Recently Used Files pane contains files that you have used previously. Double-click a file to load it.
Specifying Parameters
Click the icon “Run GSAA” to open the GSAA page. There are three categories of parameters in GSAA
- Required: Essential parameters which you must specify before the analysis can be run.
- Basic: Additional parameters with standard defaults. Typically, accepting the defaults is ok. Click Show to see these parameters.
- Advanced: Parameters that allow control of several more details of the GSAA algorithm and the java implementation. Typically, these do not need to be changed by most users. Click Show to see these parameters.
Required Fields
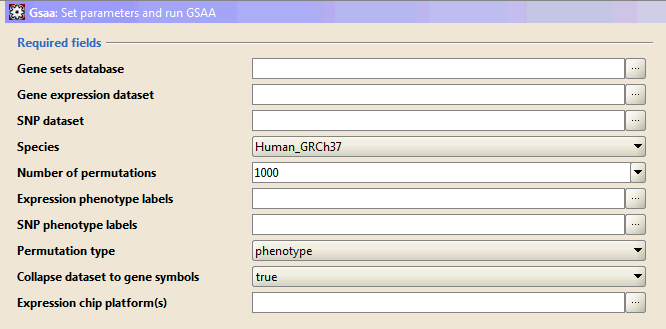
Required fields lists parameters that are essential for the analysis. Enter values for these parameters before starting the analysis.
- Gene sets database. Click the ellipse (…) button and select one or more gene sets:
- GeneMatrix (from website) lists the MSigDB gene sets available on the Broad ftp site. These gene set files may contain hundreds of gene sets. Use the Browse MSigDB Page to browse the gene sets and to create gene set files (gmx/gmt) containing only gene sets of interest.
- GeneSets(grp) lists gene sets that GSEA has created in memory; for example, gene sets created using the Text Entry tab described below.
- GeneMatrix (local gmx/gmt) lists the gene set files that you have loaded (see Loading Data).
- Subsets lists each gene set in each gmx/gmt file that you have loaded.
- Text Entry allows you to create a gene set by entering the genes for that gene set; enter one gene per line. The gene set is created in memory and deleted when you exit.
- Gene Expression dataset. Click the ellipse (…) button to select an expression dataset file from a file browser window.
- SNP dataset. Click the ellipse (…) button to select a SNP dataset file from a file browser window.
- Species. Select a species from the drop-down list. Different species use different map files for SNP-gene mapping. If you are using simulated data sets, please choose "Simulation" at the bottom of the list.
- Number of permutations. Specify the number of permutations to perform in assessing the statistical significance of the association score. It is best to start with a small number, such as 10. After the analysis completes successfully, run it again with a full set of permutations.
- Expression phenotype labels. Click the ellipse (…) button to select a phenotype labels file for expression dataset from a file browser window.
- SNP phenotype labels. Click the ellipse (…) button to select a phenotype labels file for SNP dataset from a file browser window.
- Permutation type. Select the type of permutation to perform in assessing the statistical significance of the association score:
- Phenotype. Random phenotypes are created by shuffling the phenotype labels on the samples. For each random phenotype, GSAA ranks the genes and calculates the association score for all gene sets. These association scores are used to create a null distribution from which the significance of the actual association score (for the actual expression data, SNP data and gene set) is calculated. This is the recommended method when there are at least seven (7) samples in each phenotype.
The phenotype permutation can preserve linkage disequilibrium (LD) structure in SNP data and gene-gene correlation structure in gene expression data thus it can provide a more biologically reasonable (more stringent) assessment of significance.
- Phenotype. Random phenotypes are created by shuffling the phenotype labels on the samples. For each random phenotype, GSAA ranks the genes and calculates the association score for all gene sets. These association scores are used to create a null distribution from which the significance of the actual association score (for the actual expression data, SNP data and gene set) is calculated. This is the recommended method when there are at least seven (7) samples in each phenotype.
- Collapse dataset to gene symbols:
- Select True (default) to have GSAA collapse each probe set in the expression dataset into a single vector for the gene, which gets identified by its HUGO gene symbol. When you select True, you must specify a chip annotation file (Expression chip platform parameter) and gene sets (Gene sets database parameter) that identify genes by HUGO gene symbol.
- Select False to use your expression dataset as is (with its native feature identifiers). When you select this option, the chip annotation file (Chip platform parameter) is optional and you must specify gene sets (Gene sets database parameter) that identify genes using the same feature (gene or probe) identifiers as those used in your expression dataset.
- Expression chip platform(s). Click the ellipse (…) button and select one or more DNA chip (array) annotation files for expression dataset:
- Chips (from website) lists the chip annotation files available on the Broad ftp site.
- Chips (local .chip) lists the chip annotation files that you have loaded (see Loading Data).
This parameter is mandatory or optional depending on the value of the Collapse dataset to gene symbols parameter.
Basic Fields
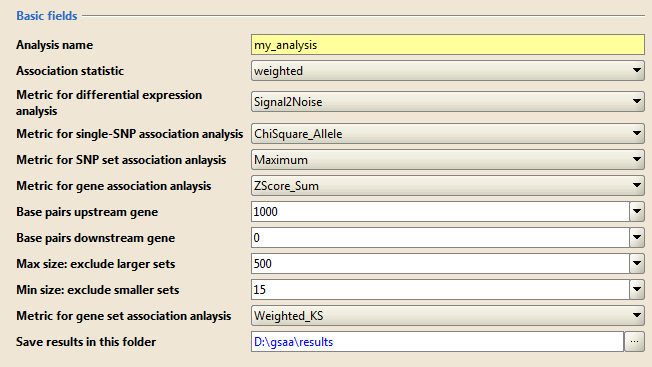
Basic fields lists additional parameters with standard defaults. Typically, you use the default values for these parameters. Click Show/Hide to display and hide these parameters.
- Analysis name. A short descriptive label for the analysis. The name cannot include spaces. This label is used as a prefix when naming the output report generated by the analysis (for example, my_analysis.Gsaa.1130510139575.rpt).
- Association statistic. This option controls the value of p used in the association score calculation: Larger p gives higher weights to genes with extreme statistic values
- classic: p=0
- weighted (default): p=1
- weighted_p2: p=2
- weighted_p1.5: p=1.5
- Metric for differential expression analysis. GSAA ranks genes by their association with phenotype and then analyzes that ranked list of genes. The association score of gene is the combination of the differential expression score and SNP set association score. Use this parameter to select the metric used to calculate the differential expression score.
Consider two phenotype classes, C1 and C2:- Signal2Noise (default) is the difference of the class means scaled by the standard deviation:
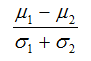
where (μ1, μ2) and (σ1, σ2) are the means and standard deviations of a gene’s expression values in classes C1 and C2, respectively. The absolute magnitude of the statistic indicates the strength of the correlation between the gene expression profile and the phenotype, and the sign indicates the direction of this correlation. - tTest is the difference of the class means scaled by the standard deviation and number of samples:
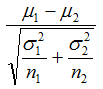
where (μ1, μ2) and (σ1, σ2) are are the means and standard deviations of a gene’s expression values in classes C1 and C2, respectively. (n1, n2) are the number of samples in classes C1 and C2. The absolute magnitude of the statistic indicates the strength of the correlation between the gene expression profile and the phenotype, and the sign indicates the direction of this correlation. - log2_Ratio_of_Classes is the log2 ratio of the class means:
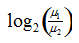
where (μ1, μ2) are the means of a gene’s expression values in classes C1 and C2, respectively. The absolute magnitude of the statistic indicates the strength of the correlation between the gene expression profile and the phenotype, and the sign indicates the direction of this correlation.
- Signal2Noise (default) is the difference of the class means scaled by the standard deviation:
- Metric for single-SNP association analysis. In SNP dataset, each gene is represented by a varied number of SNPs. GSAA assesses the association of each SNP with the phenotype and then use one of the SNP set metrics to calculate the association score of the gene in the SNP dataset. Use this parameter to select the metric used to score the SNPs in the SNP dataset.
- ChiSquare_Geno is the genotype-based chi-square score:
Suppose genotypes for a bi-allelic SNP in the SNP dataset are encoded as AA, AB, and BB, or alternatively as groups G1, G2, and G3. We first define two scaling factors, K1 and K2, to adjust for unequal sample sizes between class C1 and C2: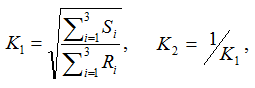
where Ri is the observed counts for group Gi in class C1, and Si is the observed counts for group Gi in class C2. The genotype-based chi-square score is then computed as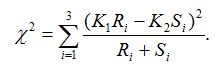
The test statistic value represents the degree to which the SNP is associated with phenotypic class distinction.
- ChiSquare_Allele is the allele-based chi-square score:
Suppose alleles for a bi-allelic SNP in the SNP dataset are encoded as A and B, or alternatively as groups G1 and G2. We first define two scaling factors, K1 and K2, to adjust for unequal sample sizes between class C1 and C2:
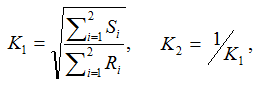
where Ri is the observed counts for group Gi in class C1, and Si is the observed counts for group Gi in class C2. The allele-based chi-square score is then computed as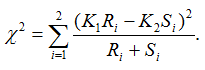
The test statistic value represents the degree to which the SNP is associated with phenotypic class distinction. - Diff_of_Major_Alleles is the absolute value of frequency difference of the major/minor allele in two classes:
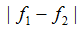
where (f1, f2) is the frequency of the major/minor allele in classes C1 and C2, respectively. The test statistic value represents the degree to which the SNP is associated with phenotypic class distinction. - tTest_Geno is a genotype-based score derived from ChiSquare_Geno:
Suppose genotypes for a bi-allelic SNP in the SNP dataset are encoded as AA, AB, and BB, or alternatively as groups G1, G2, and G3. We first define two scaling factors, K1 and K2, to adjust for unequal sample sizes between class C1 and C2:
where Ri is the observed counts for group Gi in class C1, and Si is the observed counts for group Gi in class C2. The tTest_Geno is then computed as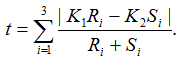
The test statistic value represents the degree to which the SNP is associated with phenotypic class distinction.
- tTest_Allele is an allele-based score derived from ChiSquare_Allele:
Suppose alleles for a bi-allelic SNP in the SNP dataset are encoded as A and B, or alternatively as groups G1 and G2. We first define two scaling factors, K1 and K2, to adjust for unequal sample sizes between class C1 and C2:
where Ri is the observed counts for group Gi in class C1, and Si is the observed counts for group Gi in class C2. The tTest_Allele is then computed as
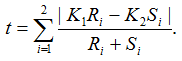
The test statistic value represents the degree to which the SNP is associated with phenotypic class distinction.
- ChiSquare_Geno is the genotype-based chi-square score:
- Metric for SNP set association analysis. In SNP dataset, each gene is represented by a varied number of SNPs. GSAA assesses the association of each SNP with the phenotype and then use one of the SNP set metrics to calculate the association score of the gene in the SNP dataset. Use this parameter to select the metric used to score the corresponding SNP set for each gene.
- Maximum (default) uses the highest association score among all SNPs mapped to the gene as the association score of that gene.
- Metric for gene association analysis. The differential gene expression score and SNP set association score for each gene are combined to generate a single gene association score. This composite correlation integrates evidence for association across the gene expression and SNP data. GSAA uses the absolute values of the differential gene expression scores for data integration across genomic sources in order to capture both up-regulation and down-regulation in pathways.
In GSAA, three methods are used to integrate the evidence from gene expression analysis and SNP analysis to produce gene association scores.
- Z-score sum
The differential expression score or SNP set association score is standardized by the mean and standard deviation of its null distribution. Suppose {e1, ..., eN} are the absolute values of differential expression scores for N genes and {s1, ..., sN} are the SNP set association scores for the same genes. The standard expression scores {ze1, ..., zeN} are computed as
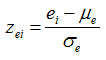
and the standard SNP set association scores {zs1, ..., zsN} for the same genes are similarly computed as
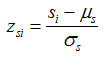
where (μe, μs) and (σe, σs) are the means and standard deviations of the null distributions corresponding to ei and si, respectively. This transformation brings the scores from different statistical tests or on different scales onto a common scale so that these scores are directly comparable with each other. The z-score transformation results in both positive values and negative values. To shift the z-scores to be positive, a constant c is added to each score. c is the absolute value of the most negative score across all standard gene expression scores and standard SNP set association scores.
The gene association scores are the sum of these standard scores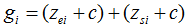
- Fisher’s method
A nominal p-value is estimated for each differential gene expression score and SNP set association score by comparing the score with its null distribution. Fisher’s method, also known as Fisher's combined probability test, is used to combine p-values from the expression-based test and the SNP-based test to produce the integrative gene association score:
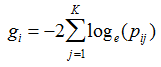
where K is the number of independent tests, in this case K=2, namely expression-based test and SNP-based test, and Pij is the p-value for gene i in test j. - Rank sum
A rank is estimated for each differential gene expression score and SNP set association score by comparing the score with its corresponding null scores. Tied values are assigned the average of the applicable ranks. For example, (2, 5, 6, 5) is ranked as (1, 2.5, 4, 2.5). Gene association scores are then computed as
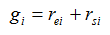
where rei and rsi are the ranks of gene i in the expression-based test and SNP-based test, respectively.
- Z-score sum
- Metric for gene set association analysis. Given the gene association scores, GSAA uses the Weighted K-S test to determine which gene sets have the greatest combined evidence for association with the given phenotype.
- Weighted Kolmogorov-Smirnov (K-S) test
The weighted K-S test determines for each gene set whether the genes belonging to that gene set are preferentially near the top of the ranked ordered list based on gene association scores. More formally, given a gene set S containing H genes and the rank ordered gene association scores {g1, ..., gN} for all genes in the data set, a running association score RASS(i) for the rank ordered genes in positions i=1, ..., N is computed as
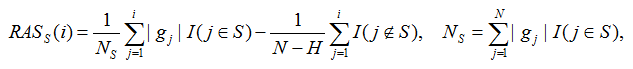
Where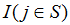 is an indicator variable that is one if the jth gene in the rank ordered list is in gene set S and otherwise zero. Similarly,
is an indicator variable that is one if the jth gene in the rank ordered list is in gene set S and otherwise zero. Similarly, 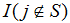 takes the value of zero if the jth gene is in the gene set and is otherwise one. The gene set association score, AS(S), is the maximum deviation from zero of the running association score over the positions i=1, ..., N
takes the value of zero if the jth gene is in the gene set and is otherwise one. The gene set association score, AS(S), is the maximum deviation from zero of the running association score over the positions i=1, ..., N
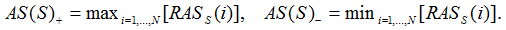
Finally, if |AS(S)+|>|AS(S)-| then the final gene set association score AS(S)=AS(S)+, otherwise AS(S)=AS(S)-. The gene association scores don’t have directionality, so the negative AS(S) means there is no association between the gene set and the phenotype. AS(S) is set to 0.0001 if AS(S)<0 so the negative AS scores will not confuse the following assignment of the direction. A same K-S test based solely on directed differential expression scores is used to get a corresponding expression-based AS score (EAS) for each gene set. The sign of the integrative AS score is then assigned to be the same as the sign of the expression-based AS score for the same gene set, AS(S)=AS(S)Xsign(EAS(S)).
- Weighted Kolmogorov-Smirnov (K-S) test
- Base pairs upstream gene. Specify the number of base pairs upstream the gene included in the SNP-gene mapping region.
- Base pairs downstream gene. Specify the number of base pairs downstream the gene included in the SNP-gene mapping region.
- Max size. After filtering from the gene sets any gene not in the expression dataset, gene sets larger than this are excluded from the analysis.
- Min size. After filtering from the gene sets any gene not in the expression dataset, gene sets smaller than this are excluded from the analysis.
- Save results in this folder. Path of the directory in which to place the analysis results. Existing results in this folder are not overwritten. By default, analysis results are saved in the GSAA output folder. To view this folder, select Help>Show GSAA output folder.
Advanced Fields
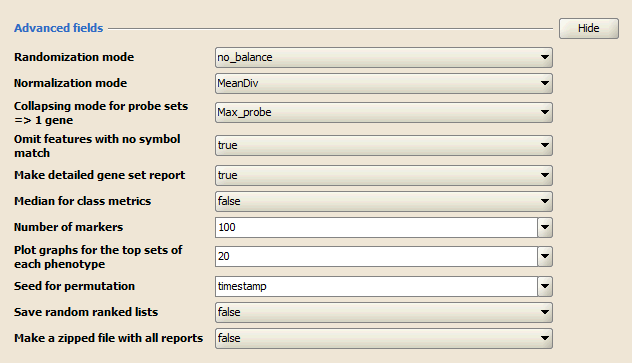
Advanced fields lists parameters that control details of the GSAA algorithm and its Java implementation. Do not change the default values of these parameters unless you are conversant with the algorithm and its Java implementation. Click Show/Hide to display and hide these parameters.
- Randomization mode. Method used to randomly assign phenotype labels to samples for phenotype permutations. Not used for gene set permutations.
- no_balance (default). Permutes labels without regard to number of samples per phenotype. For example, if your dataset has 12 samples in class_a and 10 samples in class_b, any permutation of class_a has 12 samples randomly chosen from the dataset.
- equalize_and_balance. Permutes labels by equalizing the number of samples per phenotype and then balancing the number of samples contributed by each phenotype. For example, if your dataset has 12 samples in class_a and 10 samples in class_b, any permutation of class_a has 10 samples: 5 randomly chosen from class_a and 5 randomly chosen from class_b.
We recommend using no balance (default), unless the number of samples per phenotype is highly unbalanced.
- Normalization mode. Method used to normalize the association scores (AS) across analyzed gene sets:
- MeanDiv (default): GSAA normalizes the association scores by dividing a given AS by the mean of its null distribution generated from a permutation procedure.
- None (K-S test only): GSAA does not normalize the association scores.
- Collapsing mode for probe sets => 1 gene. Used only when the Collapse dataset to gene symbols parameter is set to True. Select the expression values to use for the single probe that will represent all probe sets for the gene:
- max_probe (default): for each sample, use the maximum expression value for the probe set. For example:
Probeset_A 10 20 15 200 Probeset_B 100 105 110 95 gene_symbol_AB 100 105 110 200 - median_of_probes: for each sample, use the median expression value for the probe set.
- max_probe (default): for each sample, use the maximum expression value for the probe set. For example:
- Omit features with no symbol match. Used only when Collapse dataset to gene symbols is set to True. By default (true), the new dataset excludes probes/genes that have no gene symbols. Set to False to have the new dataset contain all probes/genes that were in the original dataset.
- Make detailed gene set report. Set to True (default) to create a detailed gene set report for each associated gene set.
- Median for class metrics. Set to True (default=False) to use the median of each class, instead of the mean, in the metrics for ranking for genes.
- Number of markers. Number of features (gene or probes) to include in the butterfly plot in the Gene Markers section of the gene set association report.
- Plot graphs for the top sets of each phenotype. Generates summary plots and detailed analysis results for the top x genes in each phenotype, where x is 20 by default. GSAA ranks gene sets by their FDR q-values so the top genes are those with the smallest FDR.
- Seed for permutation. Seed used to generate a random number for phenotype and gene set permutations: timestamp (default) or 149. The specific seed value (149) generates consistent results, which is useful when testing software.
- Save random ranked lists. Set to True (default=false) to save the random ranked lists of genes created by phenotype permutations. When you save random ranked lists, for each permutation, GSAA saves the rank metric score for each gene (the score used to position the gene in the ranked list). Saving random ranked lists is memory intensive; therefore, this parameter is set to false by default.
- Make a zipped file with all reports. Set to True (default=false) to create a zip file of the analysis results. The zip file is saved to the output folder with all of the other files generated by the analysis. This is useful for sharing analysis results.
Buttons at the bottom of the page:
- Reset. Restores the default values for all parameters.
- Last. Loads the data used the last time you ran this analysis.
- Command. Displays the command line used to run the analysis, as described in Running GSAA from the Command Line.
- Low/Normal (cpu usage). Determines the amount of CPU dedicated to this analysis. To use your computer for other tasks while running GSAA in the background, choose Low. To complete your analysis more quickly, choose Normal.
- Run. Starts the analysis.
Running Gene Set Association Analysis
![]()
Click Run to start the analysis
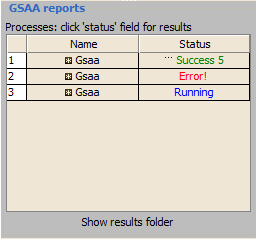
Use the Processes panel at the lower left corner to view the status of analyses run in this session, including the currently running analysis:
1. The blue Running label indicates the currently running analysis. You can click on this label to pause or resume an analysis.
2. If a red Error appears, click on it for a description of the error.
3. When the analysis completes, click the green Success label to display the results in a web browser.
Interpreting GSAA(zs-ks) Results
GSAA Statistics
Gene Set Association Score (AS)
The primary result of the gene set association analysis is the gene set association score (AS), which reflects the degree to which a gene set is overrepresented at the top of a ranked list of genes. GSAA(zs-ks) calculates the AS by walking down the ranked list of genes, increasing a running-sum statistic when a gene is in the gene set and decreasing it when it is not. The magnitude of the increment depends on the association of the gene with the phenotype. The AS is the maximum deviation from zero encountered in walking the list. A positive AS indicates gene set associated with the first phenotypic class; a negative AS indicates gene set associated with the second phenotypic class.
In the analysis results, the association plot provides a graphical view of the association score for a gene set:
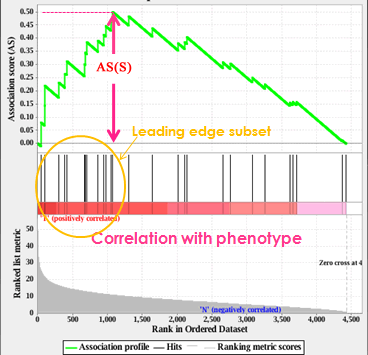
- The top portion of the plot shows the running AS for the gene set as the analysis walks down the ranked list. The score at the peak of the plot (the score furthest from 0.0) is the AS for the gene set. Since GSAA employs a non-directional differential expression score so gene sets with a distinct peak at the beginning (such as the one shown here) are generally the most interesting.
- The middle portion of the plot shows where the members of the gene set appear in the ranked list of genes.
The leading edge subset of a gene set is the subset of members that contribute most to the AS. - The bottom portion of the plot shows the value of the ranking metric as you move down the list of ranked genes. The ranking metric measures a gene’s association with a phenotype. The value of the ranking metric goes from positive to zero as you move down the ranked list.
Normalized Association Score (NAS)
By normalizing the gene set association score, GSAA accounts for differences in gene set size and in correlations between gene sets and the datasets; therefore, the normalized association scores (NAS) can be used to compare analysis results across gene sets.
Nominal P Value
GSAA uses a permutation test to evaluate the statistical significance of the AS assigned to a gene set. The statistical significance of the AS is estimated using a nominal P-value that is calculated relative to a null AS distribution generated by permutations. If the gene expression and SNP data come from the same samples, matched data, GSAA will perform better. Since it may be difficult to obtain matched genomic data and to be able to use GSAA on existing GWA and gene expression data that may not be matched we designed GSAA to allow for both matched and unmatched data. When the data are matched, permutations for the expression-based test and SNP-based test are not independent and GSAA uses the same permutation template for both. This can result in greater power to identify real associations.
False Discovery Rate (FDR) and Family-Wise Error Rate (FWER)
GSAA uses FDR and FWER to correct for multiple hypothesis testing and control the proportion of false positives below a certain threshold.
Given m gene sets {S1,S2,...,Sm} and label permutations π=1,…,Π, the FDR for each gene set Si with NAS(Si)>=0 is calculated as
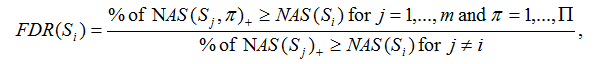
If NAS(Si)<0, the FDR is computed as
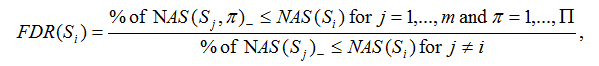
Where NAS(Sj, π) is the normalized association score for gene set j with label permutation π. NAS(Sj, π)+ and NAS(Sj, π)- denote positive and negative NAS(Sj, π), respectively. NAS(Sj) is the normalized association score for gene set j. NAS(Sj)+, NAS(Sj)- denote positive and negative NAS(Sj), respectively.
The FWER for a gene set Si with NAS(Si)>=0 is computed as
![]()
If NAS(Si)<0, the FDR is computed as
![]()
GSAA Report
This section discusses the content of the report generated by the gene set association analysis:
- Association with Phenotype
- Gene Set Details
- Gene Markers
- Other
- Detailed Association Results
- Gene Set Details Report
Association with Phenotype

The analysis report contains two “Association with Phenotype” sections. The first section shows results for gene sets that have a positive association score (gene sets that show enrichment at the top of the ranked list) and the second section shows results for gene sets that have a negative association score (gene sets that show enrichment at the bottom of the ranked list). A positive association score indicates association with the first phenotype and a negative association score indicates association with the second phenotype.
For each phenotype, the report shows:
- Number of gene sets associated with this phenotype and the total number of gene sets analyzed.
- Number of associated gene sets that are significant, as indicated by a false discovery rate (FDR) of less than 25%. Typically, these are the gene sets most likely to generate interesting hypotheses and drive further research.
- Number of associated gene sets with a nominal p value of less than 1% and of less than 5%. The nominal p value is not adjusted for gene set size or multiple hypothesis testing; therefore, it is of limited value for comparing gene sets.
- Snapshot of top results. Displays association plots for the gene sets with the smallest FDR. By default, GSAA displays plots for the top 20 gene sets. To display a different number of plots, use the Plot graphs for the top sets of each phenotype parameter on the Run GSAA Page. For a description of the association plot, see Association Score (AS).
- Detailed association results provide a summary report of gene sets associated with this phenotype (html and excel formats).
- Guide to interpret results displays this section of the documentation.
Gene Set Details

The Gene Set Details section of the analysis report provides information about the gene sets:
- Number of gene sets filtered out of the analysis due to size, and the minimum and maximum gene set sizes used for the filter.
- Number of gene sets used in the analysis.
- List of analyzed gene sets. For each gene set, the report shows the original number of genes in the gene set, the number of genes in the gene set after filtering out those genes not in the expression dataset, and the status of the gene set. Status is either blank (the gene set was included in the analysis) or “Rejected” (the gene set was filtered out of the analysis).
- The feature identifiers used for the expression dataset do not match those used in the gene sets. For example, your expression dataset contains probe identifiers from the HG_U133A chip and your gene sets identify genes based on HUGO gene symbols. For more information, see Consistent Feature Identifiers Across Data Files.
- After filtering out those genes not in the expression dataset, all of the gene sets are either larger than the maximum or smaller than the minimum gene set size allowed. You can use the Max Size and Min Size parameters on the Run GSAA Page to change the maximum and minimum gene set size.
Gene Markers

The Gene Markers section of the analysis report provides information about the ranked list of genes used for the analysis:
- Number of features (genes) in the expression dataset.
- Number of markers for each phenotype; that is, the number of genes correlated with each phenotype.
- Rank ordered list of genes in the dataset (Excel format), which includes the following information for each gene: name, p-value (from SNP-based test), gene symbol, gene title, and score (from joint analysis of gene expression and SNP genotypes).
- Heat map of the top 50 features for each phenotype and a plot showing the association between the ranked genes and the phenotypes. In a heat map, expression values are represented as colors, where the range of colors (red, pink, light blue, dark blue) shows the range of expression values (high, moderate, low, lowest).
- Butterfly plot showing the positive and negative association between gene rank and the association score (from joint analysis of gene expression and SNP genotypes). By default, the butterfly plot shows the top 100 genes; that is, the first and last 100 genes in the ranked list. You can use the Number of markers parameter on the Run GSAA Page to change the number of genes displayed.
The bottom portion of the association plot shows the observed association between gene rank and the association score for all genes in the ranked list. The butterfly plot shows the observed association, as well as permuted (1%, 5%, 50%) positive and negative associatoin, for the top genes. The butterfly plot offers one way to visualize the extent to which dataset permutations change the association between gene rank and the association score.
Other

The final section of the report, Other, lists the analysis parameters. Knowing the parameters is critical for reproducing analysis results.
Detailed Association Results
From the Association in Phenotype section of the analysis report, you can click a link to display the detailed association results report, which lists all gene sets associated with this phenotype ordered by the false discovery rate (FDR):
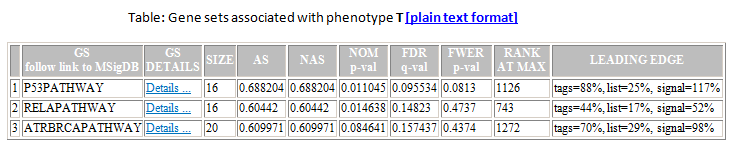
- GS. Gene set name. Click the gene set name for a detailed description of the gene set. For MSigDB gene sets, the description is the gene set page on the GSEA web site. For other gene sets, the description is provided by the author of the gene set.
- GS DETAILS. For the top 20 gene sets, click the Details link to display the Gene Set Details Report. To generate the Details link for a different number of gene sets, use the Plot graphs for the top sets of each phenotype parameter on the Run GSAA Page.
- SIZE. Number of genes in the gene set after filtering out those genes not in the expression dataset.
- AS. Association score. for the gene set; that is, the degree to which this gene set is overrepresented at the top or bottom of the ranked list of genes in the expression dataset.
- NAS. Normalized association score; that is, the association score for the gene set after it has been normalized across analyzed gene sets.
- NOM p-value. Nominal p value; that is, the statistical significance of the association score. The nominal p value is not adjusted for gene set size or multiple hypothesis testing; therefore, it is of limited use in comparing gene sets.
- FDR q-value. False discovery rate; that is, the estimated probability that the normalized association score represents a false positive finding.
- FWER p-value. Familywise-error rate; that is, a more conservatively estimated probability that the normalized association score represents a false positive finding.
- RANK AT MAX. The position in the ranked list at which the maximum association score occurred. The more interesting gene sets achieve the maximum association score near the top or bottom of the ranked list; that is, the rank at max is either very small or very large.
- LEADING EDGE. Displays the three statistics used to define the leading edge subset:
- Tags. The percentage of gene hits before (for positive AS) or after (for negative AS) the peak in the running association score. This gives an indication of the percentage of genes contributing to the association score.
- List. The percentage of genes in the ranked gene list before (for positive AS) or after (for negative AS) the peak in the running association score. This gives an indication of where in the list the association score is attained.
- Signal. The association signal strength that combines the two previous statistics:
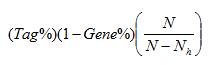
where N is the number of genes in the list and Nh is the number of genes in the gene set. If the gene set is entirely within the first Nh positions in the list, then the signal strength is maximal or 100%. If the gene set is spread throughout the list, then the signal strength decreases towards 0%.
These statistics describe the leading-edge subset of a single gene set. Use the Leading Edge analysis to analyze the overlap between multiple leading-edge subsets.
Gene Set Details Report
From the Detailed Association Results table, click the Details link for a gene set to display a Gene Set Details report that contains the following:
- A table showing the GSAA results for this gene set. The fields in this table are similar to those in the Detailed Association Results.
- An association plot for this gene set, as described in Association Score (AS).
- A table of genes in the gene set ordered by their position in the ranked list of genes. The analysis includes only those genes in the gene set that are also in the expression dataset. To display the table in Excel, click the plain text format link in the table header.
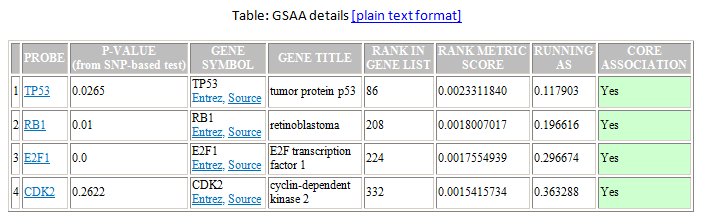
- PROBE. Probe used for the gene. When possible, the probe name links to probe information.
- P-VALUE. P-value of rank metric score of gene in SNP based test.
- GENE SYMBOL. Gene name. If you specify a chip annotation file, the report includes the gene symbol name with links to external databases that provide gene information.
- GENE TITLE. Brief description of the gene from the chip annotation file.
- RANK IN GENE LIST. Position of the gene in the ranked list of genes.
- RANK METRIC SCORE. Score used to position the gene in the ranked list.
- RUNNING AS. Running association score; that is, the association score at this point in the ranked list of genes.
- CORE ASSOCIATION . Genes with a Yes value in this column contribute to the leading-edge subset within the gene set. This is the subset of genes that contributes most to the association result. Use the Leading Edge analysis to analyze the overlap between multiple leading-edge subsets.
- A heat map of the genes in the gene set. In a heat map, expression values are represented as colors, where the range of colors (red, pink, light blue, dark blue) shows the range of expression values (high, moderate, low, lowest).
- A histogram of the association scores for all permutations.
Running GSAA from the Command Line
Syntax
To run GSAA from the command line, use a java command of the form:
java -cp full-path/GSAA.jar –Xmx5000m gsaa-tool parameters
- -cp Points the CLASSPATH variable to the complete path of the GSAA.jar file. You do not need to set any other CLASSPATH variables.
- -Xmx1000m Specifies the amount of memory available to Java. GSAA has been successfully used with 20000m on a Linux server for a large GWA dataset and 10000 permutations of phenotype labels.
- gsaa-tool Specifies the analysis to use. For GSAA, use xtools.gsea.Gsaa; for GSAA-SNP, use xtools.gsea.GsaaSnp.
- parameters Specifies the analysis parameters. To find the parameters for an analysis, open the GSAA application, display the page that runs the analysis, enter the parameters that you want to use, and click the Command button at the bottom of the page. GSAA displays the command line used to run the analysis. If you omit a parameter, GSAA uses the default value as displayed in the GSAA application.
- Paths to file names must be fully specified or relative to the execution directory. When creating batch files, you generally want to use full path names for all files.
- File names are platform-specific and may require editing. For example, on Windows, a file name that contains spaces must be enclosed in quotation marks.
- Files cannot be directly accessed from the GSEA ftp site. Download the desired gene set or array annotations files from the GSEA web site (http://www.broad.mit.edu/gsea/downloads.jsp) and reference the downloaded files in the command line.
- Parameter values cannot include hyphens (-); therefore, file names cannot include hyphens. If necessary, change hyphens to underscores. For example, you cannot use -res my-dataset.gct, but must use -res my_dataset.gct instead.
Parameters
The table below lists the command line options and their corresponding names in the graphical user interface (GUI).
| Command line option | GUI name |
|---|---|
| -gmx | Gene sets database |
| -exp_file | Gene expression dataset |
| -snp_file | SNP dataset |
| -exp_template_file | Expression phenotype labels |
| -snp_template_file | SNP phenotype labels |
| -species | Species |
| -permute | Permutation type |
| -rnd_type | Randomization mode |
| -nperm | Number of permutations |
| -scoring_scheme | Association statistic |
| -collapse | Collapse dataset to gene symbols |
| -norm | Normalization mode |
| -mode | Collapsing mode for probe sets=>1 gene |
| -rpt_label | Analysis name |
| -metric | Metric for differential gene expression anlaysis |
| -cmetric | Metric for single-SNP association analysis |
| -chip | Expression chip platform(s) |
| -cmetric_snpset | Metric for SNP set association analysis |
| -imetric | Metric for gene association analysis |
| -include_only_symbols | Omit features with no symbol match |
| -make_sets | Make detailed gene set report |
| -median | Median for class metrics |
| -num | Number of markers |
| -plot_top_x | Plot graphs for the top sets of each phenotype |
| -rnd_seed | Seed for permutation |
| -save_rnd_lists | Save random ranked lists |
| -set_a_upstream | Base pairs upstream gene |
| -set_b_downstream | Base pairs downstream gene |
| -set_max | exclude larger sets |
| -set_min | exclude smaller sets |
| -smetric | Metric for gene set association analysis |
| -zip_report | Make a zipped file with all reports |
| -out | save results in this folder |
| -gui | graphical user interface |
Examples
1, Following is a command line that assumes that you use the in-house gene set collection in GSAA, click HERE to see a list of available gene sets databases.
java -cp /home/tcga/program/GSAA.jar -Xmx10000m xtools.gsea.Gsaa -gmx Genesets:MSigDB.c2.cp.v5.0.symbols.gmt -exp_file /home/tcga/data/ gbm_tcga.gct -snp_file /home/tcga/data/gbm_tcga.snp -exp_template_file /home/tcga/data/exp_gbm_tcga.cls -snp_template_file /home/tcga/data/snp_gbm_tcga.cls -species Human -permute phenotype -rnd_type no_balance -nperm 10000 -scoring_scheme weighted -collapse false -norm MeanDiv -mode Max_probe -rpt_label tcga_10000_c2.cp.v5.0 -metric Signal2Noise -cmetric ChiSquare_Allele -chip gseaftp.broadinstitute.org://pub/gsea/annotations/HG_U133A.chip -cmetric_snpset Maximum -imetric ZScore_Sum -include_only_symbols true -make_sets true -median false -num 100 -plot_top_x 20 -rnd_seed timestamp -save_rnd_lists false -set_a_upstream 1000 -set_b_downstream 1000 -set_max 100 -set_min 15 -smetric Weighted_KS -zip_report false -out /home/tcga/result -gui false
2, Following is a command line that assumes that you supply the gene sets database file and chip annotation file.
java -cp /home/tcga/program/GSAA.jar -Xmx10000m xtools.gsea.Gsaa -gmx /home/tcga/data/c2.cp.v5.0.symbols.gmt -exp_file /home/tcga/data/ gbm_tcga.gct -snp_file /home/tcga/data/gbm_tcga.snp -exp_template_file /home/tcga/data/exp_gbm_tcga.cls -snp_template_file /home/tcga/data/snp_gbm_tcga.cls -species Human -permute phenotype -rnd_type no_balance -nperm 10000 -scoring_scheme weighted -collapse false -norm MeanDiv -mode Max_probe -rpt_label tcga_10000_c2.cp.v5.0 -metric Signal2Noise -cmetric ChiSquare_Allele -chip /home/tcga/data/HG_U133A.chip -cmetric_snpset Maximum -imetric ZScore_Sum -include_only_symbols true -make_sets true -median false -num 100 -plot_top_x 20 -rnd_seed timestamp -save_rnd_lists false -set_a_upstream 1000 -set_b_downstream 1000 -set_max 100 -set_min 15 -smetric Weighted_KS -zip_report false -out /home/tcga/result -gui false